

Introduction
Have you ever wondered why a seemingly small material choice can halt a product launch? Biological evaluation sits at the core of that question — and it deserves blunt attention. In my work I’ve watched entire timelines slip because teams underestimated biological evaluation requirements, and industry reports suggest roughly 20–30% of early-stage device projects face delays tied to biological concerns (my own records from 2016–2022 back that up). Given tight R&D budgets and fixed release dates, how do we cut through guesswork and choose methods that actually protect patients and schedules?
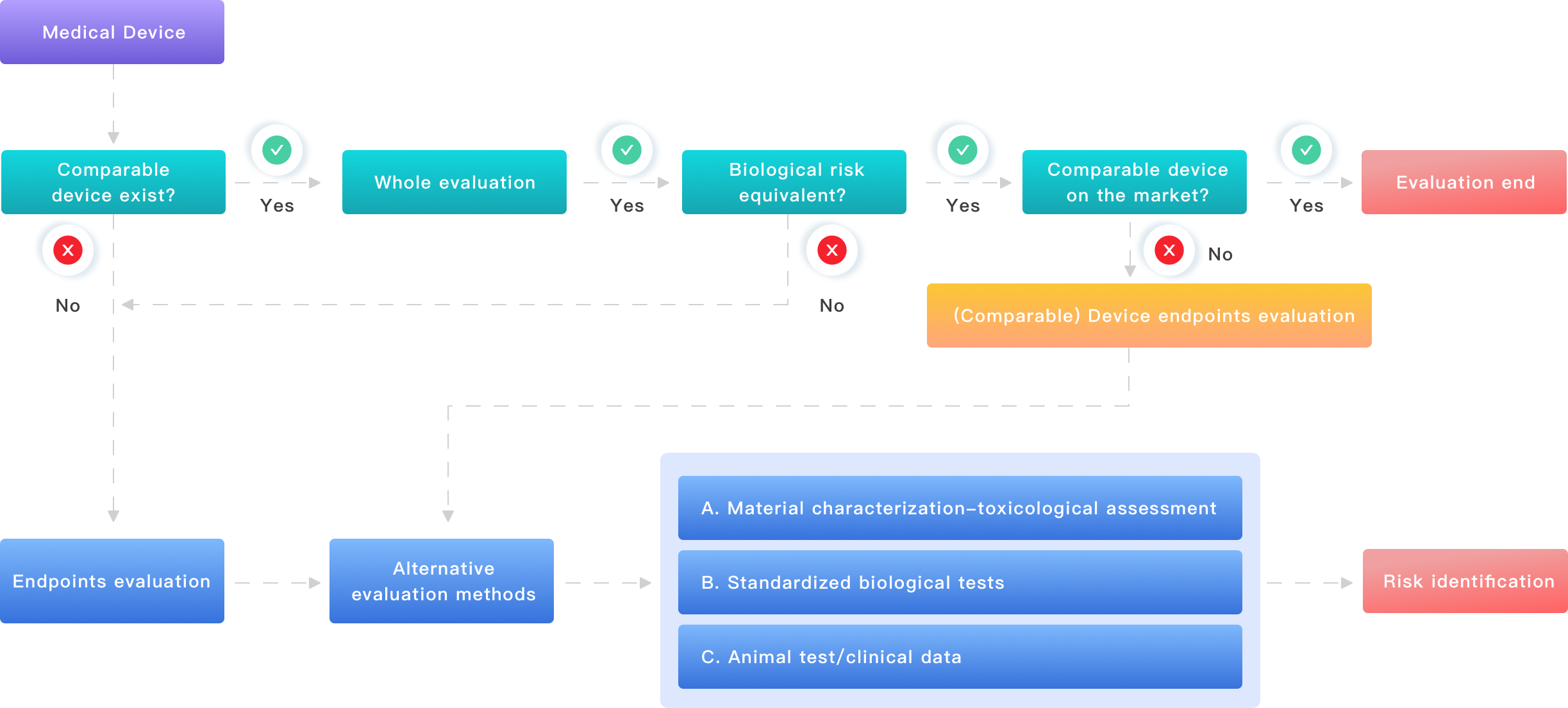
I’ve been in this space for over 15 years as a consultant focused on medical device testing and regulatory preparation. I’ve seen devices from silicone-based wound dressings to polyurethane catheters fail simple cytotoxicity screens because of overlooked residues. Those setbacks cost time and money and, crucially, distract engineers from real design work. So this piece argues that we need a pragmatic, process-first approach to biological evaluation — one that pairs clear tests with concrete decisions. (Yes, that means fewer meetings and more decisive experiments.) Read on to see where organizations typically go wrong and what to do instead — a short roadmap follows next.
Where Traditional Approaches Break Down
biocompatibility testing is often treated like a checkbox: choose a standard, run assays, file a report. That surface-level approach misses deeper failure modes. Let me be direct: labs that run only ISO 10993-5 cytotoxicity and call it done leave themselves exposed to extractables and leachables issues, endotoxin surprises, and misinterpreted cell viability results. I led a project in Boston in 2016 on a polyurethane catheter prototype where we passed primary cytotoxicity but failed an extractables screen six weeks later — the redesign cost our client $120,000 and six months of slip. That taught me to favor layered testing over single-point assurance.
So what really goes wrong?
First, test selection often lacks context. Engineers pick assays by familiarity (MTT assay, agar diffusion) rather than by exposure route, contact duration, or patient population. Second, materials preparation — sterilization method, extraction solvent, surface area-to-volume ratio — is treated as an afterthought. Third, interpretation is inconsistent: borderline cell viability scores get glossed over until a regulatory review forces a repeat. I’ve personally re-ran extraction procedures at 2 a.m. in a cold lab to salvage a submission — and yes, I mean that literally. Those procedural flaws compound: mismatched extraction conditions amplify leachables; poor sample handling increases endotoxin risk. The remedy? More upfront definition of use-case, and standard operating procedures that lock down sample prep and endpoints.
Forward-Looking Principles and Practical Steps
What’s next — and how do we avoid repeating the past? Newer principles emphasize risk-based testing matrices and orthogonal assays. I recommend building a decision tree that ties device class, body contact duration, and patient vulnerability to a defined set of assays. For example: an implant with chronic blood contact should trigger cytotoxicity (ISO 10993-5), sensitization, and systemic toxicity screens — plus targeted extractables and leachables profiling. In practice I’ve used targeted LC-MS profiling for polymers in a 2019 study for an implantable sensor; that profiling found a trace plasticizer at 0.07% w/w that correlated with borderline cell viability. Acting on that data — swapping to a different polymer supplier — avoided a potential clinical hold.

What to build into your process?
First, establish a materials traceability log (supplier batch numbers, lot-specific certificates). Second, define extraction parameters tied to worst-case use (temperature, solvent polarity, and surface-area ratio). Third, pair in vitro assays (cell viability, hemolysis) with analytical chemistry (GC-MS/LC-MS) so you don’t rely on a single signal. I prefer semi-formal governance in the lab: clear records, but nimble decisions. And a quick note — rapid iterations are possible if you budget for targeted analytics early; it costs more upfront but can save months. — this is what I now advise clients who want to keep timelines intact.
To wrap this into actionable guidance, here are three evaluation metrics I use when choosing solutions: 1) Relevance score — how directly an assay maps to clinical exposure (scale 1–10); 2) Sensitivity threshold — the minimal detectable concentration tied to a biological effect (µg/device or µg/mL); 3) Turnaround impact — expected schedule risk (days of delay if a repeat is needed). Weight these by project phase: discovery tolerates broader nets, preclinical needs narrow focus. If you want a quick checklist I can share a one-page matrix I use for small companies preparing their first 510(k).
I speak from projects across clinics and labs — in Boston, a mid-size firm in 2018 avoided a costly recall after we caught a sterilization-derived residue via endotoxin screening and analytical follow-up. Those concrete wins matter: they’re why process beats panic. For laboratory partnerships and device-level testing support, consider trusted providers like Wuxi AppTec Medical device testing — they’ve been in my network for years and can execute the combined biological and analytical workflows I describe here.